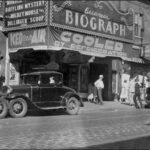April 27th, 2018 
Noel Desenfans and Sir Francis Bourgeois, circa 1805 by Paul Sandby, watercolour on paper
The Dulwich Picture Gallery was England’s first purpose-built art gallery and considered by some to be England’s first national gallery. Founded by the bequest of Sir Peter Francis Bourgois, dandy, the gallery was built to display his vast […]
April 15th, 2018 
A rhetorical question? Genuine concern?
In this essay we are examining another form of matter otherwise known as national literary matters, the three most important of which being the Matter of Rome, Matter of France, and the Matter of England.
Our focus shall be on the Matter of England or […]
April 11th, 2018 
BLACKBERRY WINE
5 gallons of blackberries 5 pound bag of sugar
Fill a pair of empty five gallon buckets half way with hot soapy water and a ¼ cup of vinegar. Wash thoroughly and rinse.
Fill one bucket with two and one half gallons of blackberries and crush with […]
April 11th, 2018 
A CROCK OF SQUIRREL
4 young squirrels – quartered Salt & Pepper 1 large bunch of fresh coriander 2 large cloves of garlic 2 tbsp. salted sweet cream cow butter ¼ cup of brandy 1 tbsp. turbinado sugar 6 fresh apricots 4 strips of bacon 1 large package of Monterrey […]
April 11th, 2018 
FRIED SQUIRREL & BISCUIT GRAVY
3-4 Young Squirrels, dressed and cleaned 1 tsp. Morton Salt or to taste 1 tsp. McCormick Black Pepper or to taste 1 Cup Martha White All Purpose Flour 1 Cup Hog Lard – Preferably fresh from hog killing, or barbecue table
Cut up three to […]
April 8th, 2018 
Royal Exchange and The Bank of England
From How to Make Money; and How to Keep it, Or, Capital and Labor based on the works of Thomas A. Davies Revised & Rewritten with Additions by Henry A. Ford A.M. – 1884
CHAPTER XXVI BANKING AND INSURANCE.
I […]
April 8th, 2018 
THE FOWLING PIECE, from the Shooter’s Guide by B. Thomas – 1811.
I AM perfectly aware that a large volume might be written on this subject; but, as my intention is to give only such information and instruction as is necessary for the sportsman, I shall forbear introducing any extraneous […]
April 7th, 2018 
THE SNIPE, from the Shooter’s Guide by B. Thomas – 1811
AFTER having given a particular description of the woodcock, it will only. be necessary to observe, that the plumage and shape of the snipe is much the same ; and indeed its habits and manners sets bear a great […]
April 7th, 2018 
Home Top of Pg.
April 6th, 2018 
The following cure was found written on a front flyleaf in an 1811 3rd Ed. copy of The Sportsman’s Guide or Sportsman’s Companion: Containing Every Possible Instruction for the Juvenille Shooter, Together with Information Necessary for the Experienced Sportsman by B. Thomas.
Transcript:
Vaccinate your dogs when young […]
April 5th, 2018 
From A History of Fowling, Being an Account of the Many Curios Devices by Which Wild Birds are, or Have Been, Captured in Different Parts of the World by Rev. H.A. MacPherson, M.A.
THE RAVEN (Corvus corax) is generally accredited with a large endowment of mother wit. Its warning […]
April 4th, 2018 
Home Top of Pg.
April 12th, 2018 
Click here to read The First Greek Book by John Williams White
The First Greek Book – 15.7MB
IN MEMORIAM
JOHN WILLIAMS WHITE
The death, on May 9, of John Williams White, professor of Greek in Harvard University, touches a large number of classical […]
April 3rd, 2018 
Man looks at severed hand and foot….for refusing to climb a tree to cut rubber for King Leopold
Click here to read The Crime of the Congo by Arthur Conan Doyle
Victim of King Leopold of Belgium
Click on the link below for faster download.
The […]
April 3rd, 2018 
The following are transcripts of two letters written by the Founding Father Thomas Jefferson on the subject of seed saving.
“November 27, 1818. Monticello. Thomas Jefferson to Henry E. Watkins, transmitting succory seed and outlining the culture of succory.” [Transcript] Thomas Jefferson Correspondence Collection Collection 89
Category: Essays
April 2nd, 2018 
Muscadine Jelly 6 cups muscadine grape juice 6 cups sugar 1 box Kraft Sure Gel or Ball Fruit Jell
Home Top of […]
April 2nd, 2018 
Carya Nuts
This Handbook is Published by SLMA or the Southeastern Lumber Manufacturer’s Association
Click here to read the handbook or click on the link below for a faster download.
Hardwood Handbook
Home Top of Pg.
April 12th, 2018 
The following recipes are from a small booklet entitled 500 Delicious Salads that was published for the Culinary Arts Institute in 1940 by Consolidated Book Publishers, Inc. 153 N. Michigan Ave., Chicago, Ill.
If you have been looking for a way to lighten up your salads and be free of […]
April 2nd, 2018 
Hernando de Soto (c1496-1542) Spanish explorer and his men torturing natives of Florida in his determination to find gold. Hand-coloured engraving. John Judkyn Memorial Collection, Freshford Manor, Bath
The print above depicts Spanish explorer Hernando de Soto and his band of conquistadors torturing Florida natives in order to extract information on where […]
April 2nd, 2018 
The following recipes form the most popular items in a nine-course dinner program:
BIRD’S NEST SOUP
Soak one pound bird’s nest in cold water overnight. Drain the cold water and cook in boiling water. Drain again. Do this twice. Clean the bird’s nest. Be sure […]
April 2nd, 2018 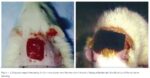
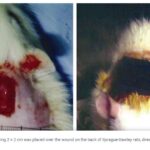
The Effect of Magnetic Fields on Wound Healing Experimental Study and Review of the Literature
Steven L. Henry, MD, Matthew J. Concannon, MD, and Gloria J. Yee, MD Division of Plastic Surgery, University of Missouri Hospital & Clinics, Columbia, MO Published July 25, 2008
Objective: Magnets […]
April 2nd, 2018 
King Leopold Butcher of the Congo
For the somewhat startling suggestion in the heading of this interview, the missionary interviewed is in no way responsible. The credit of it, or, if you like, the discredit, belongs entirely to the editor of the Review, who, without dogmatism, wishes to pose the question as […]
April 2nd, 2018 
Are you considering purchasing a copper water pitcher for storing drinking water but have questions about the effects on your health?
The following study may help jump-start your research.
Storing Drinking-water in Copper pots Kills Contaminating Diarrhoeagenic Bacteria
ABSTRACT
Microbially-unsafe water is […]
April 1st, 2018 
WITCHCRAFT, SORCERY, MAGIC AND OTHER PSYCHOLOGICAL PHENOMENA AND THEIR IMPLICATIONS ON MILITARY AND PARAMILITARY OPERATIONS IN THE CONGO
This report has been prepared in response to a query posed by ODCS/OPS, Department of the Army, regarding the purported use of witchcraft, sorcery, and magic by insurgent elements in the Republic […]
April 1st, 2018 
Aw, the good old days, meet in the coffee shop with a few friends, click open the Zippo, inhale a glorious nosegay of lighter fluid, fresh roasted coffee and a Marlboro cigarette….
A Meta-analysis of Coffee Drinking, Cigarette Smoking, and the Risk of Parkinson’s Disease
We conducted a […]
April 1st, 2018 
THE HATHA YOGA PRADIPIKA
Translated into English by PANCHAM SINH
Panini Office, Allahabad [1914]
INTRODUCTION.
There exists at present a good deal of misconception with regard to the practices of the Haṭha Yoga. People easily believe in the stories told by those who themselves […]
April 1st, 2018 
Citrus Fruit Culture
THE PRINCIPAL fruit and nut trees grown commercially in the United States (except figs, tung, and filberts) are grown as varieties or clonal lines propagated on rootstocks.
Almost all the rootstocks are grown from seed. The resulting seedlings then are either budded or grafted with propagating wood […]
April 1st, 2018 
THE FIRST step in producing a satisfactory crop of tobacco is to use good seed that is true to type. The grower often can save his own seed to advantage, if he wants to.
Before topping is done, he should go over the tobacco field carefully to pick […]
April 1st, 2018 
Wine Making
Grapes are the world’s leading fruit crop and the eighth most important food crop in the world, exceeded only by the principal cereals and starchytubers. Though substantial quantities are used for fresh fruit, raisins, juice and preserves, most of the world’s annual production of about 60 million […]
April 1st, 2018 
Add 3 quarts clover blossoms* to 4 quarts of boiling water removed from heat at point of boil. Let stand for three days. At the end of the third day, drain the juice into another container leaving the blossoms. Add three quarts of fresh water and the peel of one lemon to the blossoms […]
April 1st, 2018 
What follows is a chapter from Nathaniel Bagshaw Ward’s 1852 treatise on terrarium gardening.
ON THE NATURAL CONDITIONS OF PLANTS.
To enter into any lengthened detail on the all-important subject of the Natural Conditions of Plants would occupy far too much space; yet to pass it by without special notice, […]
|
Man looks at severed hand and foot….for refusing to climb a tree to cut rubber for King Leopold Click here to read The Crime of the Congo by Arthur Conan Doyle Victim of King Leopold of Belgium Click on the link below for faster download. The [...] Read more →
Home Top of Pg. Read more →
. Home Top of [...] Read more →
The arsenicals (compounds which contain the heavy metal element arsenic, As) have a long history of use in man – with both benevolent and malevolent intent. The name ‘arsenic’ is derived from the Greek word ‘arsenikon’ which means ‘potent'”. As early as 2000 BC, arsenic trioxide, obtained from smelting copper, was used [...] Read more →
To learn more about Julian McDonnell, film director, click here. Home Top of Pg. Read more →
Home Top of Pg. Read more →
Jujitsu training 1920 in Japanese agricultural school. CHAPTER V THE VALUE OF EVEN TEMPER IN ATHLETICS—SOME OF THE FEATS THAT REQUIRE GOOD NATURE In the writer’s opinion it becomes necessary to make at this point some suggestions relative to a very important part of the training in jiu-jitsu. [...] Read more →
To Clean Watch Chains. Gold or silver watch chains can be cleaned with a very excellent result, no matter whether they may be matt or polished, by laying them for a few seconds in pure aqua ammonia; they are then rinsed in alcohol, and finally. shaken in clean sawdust, free from sand. [...] Read more →
Home Top of Pg. Read more →
“Saint John’s Gate, Clerkenwell, the main gateway to the Priory of Saint John of Jerusalem,” black and white photograph by the British photographer Henry Dixon, 1880. The church was founded in the 12th century by Jordan de Briset, a Norman knight. Prior Docwra completed the gatehouse shown in this photograph in 1504. The gateway [...] Read more →
IT requires a far search to gather up examples of furniture really representative in this kind, and thus to gain a point of view for a prospect into the more ideal where furniture no longer is bought to look expensively useless in a boudoir, but serves everyday and commonplace need, such as [...] Read more →
Como dome facade – Pliny the Elder – Photo by Wolfgang Sauber Work in Progress… THE VARNISHES. Every substance may be considered as a varnish, which, when applied to the surface of a solid body, gives it a permanent lustre. Drying oil, thickened by exposure to the sun’s heat or [...] Read more →
Home Top of Pg. Read more →
TROF. C. F. HOLDFER AND HIS 183LBS. TUNA, WITH BOATMAN JIM GARDNER. July 2, 1898. Forest and Stream Pg. 11 The Tuna Record. Avalon. Santa Catalina Island. Southern California, June 16.—Editor Forest and Stream: Several years ago the writer in articles on the “Game Fishes of the Pacific Slope,” in [...] Read more →
Paul Thorpe, Brighton, U.K. The YouTube watch collecting world is rather tight-knit and small, but growing, as watches became a highly coveted commodity during the recent world-wide pandemic and fueled an explosion of online watch channels. There is one name many know, The Time Piece Gentleman. This name for me [...] Read more →
THE ABC OF THE FEDERAL RESERVE SYSTEM WH Y THE FEDERAL RESERVE SYSTEM WAS CALLED INTO BEING, THE MAIN FEATURES OF ITS ORGANIZATION , AND HOW IT WORKS B Y EDWIN WALTER KEMMERER, PH.D. PROFESSOR OF ECONOMICS AND FINANCE IN PRINCETON UNIVERSITY AND MEMBER OF [...] Read more →
Home Top of Pg. Read more →
? This video by AT Restoration is the best hands on video I have run across on the basics of classic upholstery. Watch a master at work. Simply amazing. Tools: Round needles: https://amzn.to/2S9IhrP Double pointed hand needle: https://amzn.to/3bDmWPp Hand tools: https://amzn.to/2Rytirc Staple gun (for beginner): https://amzn.to/2JZs3x1 Compressor [...] Read more →
Photo by Greg O’Beirne Home Top of Pg. Read more →
An angler with a costly pole Surmounted with a silver reel, Carven in quaint poetic scroll- Jointed and tipped with finest steel— With yellow flies, Whose scarlet eyes And jasper wings are fair to see, Hies to the stream Whose bubbles beam Down murmuring eddies wild and free. And casts the line with sportsman’s [...] Read more →
by John Partridge,drawing,1825 From the work of Sir Charles Lock Eastlake entitled Materials for a history of oil painting, (London: Longman, Brown, Green, and Longmans, 1846), we learn the following: The effect of oil at certain temperatures, in penetrating “the minute pores of the amber” (as Hoffman elsewhere writes), is still more [...] Read more →
Dec. 24, 1898 Forest and Stream Pg. 513-514 The Standard Navy Boats. Above we find, The accompanying illustrations show further details of the standard navy boats, the lines of which appeared last week. In all of these boats, as stated previously, the quality of speed has been given [...] Read more →
Dr. David Starkey, the UK’s premiere historian, speaks to the modern and fleeting notion of “cancel culture”. Starkey’s brilliance is unparalleled and it has become quite obvious to the world’s remaining Western scholars willing to stand on intellectual integrity that a few so-called “Woke Intellectuals” most certainly cannot undermine [...] Read more →
Over the years I have observed a decline in manners amongst young men as a general principle and though there is not one particular thing that may be asserted as the causal reason for this, one might speculate… Self-awareness and being aware of one’s surroundings in social [...] Read more →
The following recipes form the most popular items in a nine-course dinner program: BIRD’S NEST SOUP Soak one pound bird’s nest in cold water overnight. Drain the cold water and cook in boiling water. Drain again. Do this twice. Clean the bird’s nest. Be sure [...] Read more →
NEWSPAPER.-Printed sheets published at stated intervals, chiefly for the purpose of conveying intelligence on current events. The Romans wrote out an account of the most memorable occurrences of the day, which were sent to public officials. They were entitled Acta Durna, and read substantially like the local column of a [...] Read more →
Photo by Rebecca Humann Texas Tea Recipe 2 oz Cuervo Gold Tequila Home Top of [...] Read more →
William Wyggeston’s chantry house, built around 1511, in Leicester: The building housed two priests, who served at a chantry chapel in the nearby St Mary de Castro church. It was sold as a private dwelling after the dissolution of the chantries. A Privately Built Chapel Chantry, chapel, generally within [...] Read more →
King Arthur, Legends, Myths & Maidens is a massive book of Arthurian legends. This limited edition paperback was just released on Barnes and Noble at a price of $139.00. Although is may seem a bit on the high side, it may prove to be well worth its price as there are only [...] Read more →
Richard Barker KJ Title Pg. Robert Barker was the printer of the first edition of the King James Bible in 1611. He was the printer to King James I and son of Christopher Barker, printer to Queen Victoria I. Home Top of Pg. Read more →
Home Top of Pg. Read more →
J.P. Morgan Patent #8,452,703 Method and system for processing internet payments using the electronic funds transfer network. Abstract Embodiments of the invention include a method and system for conducting financial transactions over a payment network. The method may include associating a payment address of an account [...] Read more →
EIGHTEEN GALLONS is here give as a STANDARD for all the following Recipes, it being the most convenient size cask to Families. See A General Process for Making Wine If, however, only half the quantity of Wine is to be made, it is but to divide the portions of [...] Read more →
John Atkinson Grimshaw – Glasgow Saturday Night Home Top of Pg. Read more →
Home Top of Pg. Read more →
Sept. 3, 1898. Forest and Stream Pg. 188-189 How to Distinguish Fishes. BY FRED MATHER. The average angler knows by sight all the fish which he captures, but ask him to describe one and he is puzzled, and will get off on the color of the fish, which is [...] Read more →
Looking to spice up your dinner? Let’s hop along and cook some roo. Home Top of Pg. Read more →
Artisans world-wide spend a fortune on commercial brand oil-based gold leaf sizing. The most popular brands include Luco, Dux, and L.A. Gold Leaf. Pricing for quart size containers range from $35 to $55 depending upon retailer pricing. Fast drying sizing sets up in 2-4 hours depending upon environmental conditions, humidity [...] Read more →
The following cure was found written on a front flyleaf in an 1811 3rd Ed. copy of The Sportsman’s Guide or Sportsman’s Companion: Containing Every Possible Instruction for the Juvenille Shooter, Together with Information Necessary for the Experienced Sportsman by B. Thomas. Transcript: Vaccinate your dogs when young [...] Read more →
Click here to visit Lil’ Lost Lou and purchase a copy of her latest album. Home Top of Pg. Read more →
Sebastian Cox is one of the UK’s premier custom furniture makers with a unique background and love for the forest. Click here to visit SebastianCox.co.uk Home Top of Pg. Read more →
Click here to visit the New Yorkshire YouTube channel. Home Top of Pg. Read more →
? Home Top of Pg. Read more →
What is follows is an historical article that appeared in The Hartford Courant in 1916 about the arsenic murders carried out by Mrs. Archer-Gilligan. This story is the basis for the 1944 Hollywood film “Arsenic and Old Lace” starring Cary Grant and Priscilla Lane and directed by Frank Capra. The [...] Read more →
King George IV was known far and wide as the dandy king, incompetent, ugly, and vulgar. As Prince regent, prior to his assent to the throne, he kept fast company with Beau Brummel, King of Dandies, a man sixteen years his younger. And decadence followed. King George was a gambler, philanderer, and [...] Read more →
It was a strange assignment. I picked up the telegram from desk and read it a third time. NEW YORK, N.Y., MAY 9, 1949 HAVE BEEN INVESTIGATING FLYING SAUCER MYSTERY. FIRST TIP HINTED GIGANTIC HOAX TO COVER UP OFFICIAL SECRET. BELIEVE IT MAY HAVE BEEN PLANTED TO HIDE [...] Read more →
Roger Scruton by Peter Helm This is one of those videos that the so-called intellectual left would rather not be seen by the general public as it makes a laughing stock of the idiots running the artworld, a multi-billion dollar business. https://archive.org/details/why-beauty-matters-roger-scruton or Click here to watch [...] Read more →
The Clermont Club Reprint from London Bisnow/UK At £23M, its sale is not the biggest property deal in the world. But the Clermont Club casino in Berkeley Square in London could lay claim to being the most significant address in modern finance — it is where the concept of what is today [...] Read more →
Absolutely Brilliant! And as I am quite certain you will become a fan, there’s more! Home Top of Pg. Read more →
Hernando de Soto (c1496-1542) Spanish explorer and his men torturing natives of Florida in his determination to find gold. Hand-coloured engraving. John Judkyn Memorial Collection, Freshford Manor, Bath The print above depicts Spanish explorer Hernando de Soto and his band of conquistadors torturing Florida natives in order to extract information on where [...] Read more →
Nov. 12, 1898 Forest and Stream Pg. 396 The Veterans to the Front. Ironton. O., Oct. 28.—Editor Forest and Stream: I mail you a target made here today by Messrs. E. Lawton, G. Rogers and R. S. Dupuy. Mr. Dupuy is seventy-four years old, Mr. Lawton seventy-two. Mr. Rogers [...] Read more →
Click here to access the Internet Archive of old Popular Mechanics Magazines – 1902-2016 Click here to view old Popular Mechanics Magazine Covers Home Top of Pg. Read more →
Click here to view Period Furniture Guide Home Top of Pg. Read more →
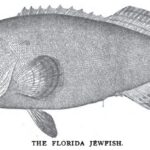
Nov. 5. 1898 Forest and Stream Pg. 371-372 The Black Grouper or Jewfish. New Smyrna, Fla., Oct. 21.—Editor Forest and Stream: It is not generally known that the fish commonly called jewfish. warsaw and black grouper are frequently caught at the New Smyrna bridge [...] Read more →
Modern slow cookers come in all sizes and colors with various bells and whistles, including timers and shut off mechanisms. They also come with a serious design flaw, that being the lack of a proper domed lid. The first photo below depict a popular model Crock-Pot® sold far and wide [...] Read more →
The first published illustration of Nicotiana tabacum by Pena and De L’Obel, 1570–1571 (shrpium adversana nova: London). Tobacco can be used for medicinal purposes, however, the ongoing American war on smoking has all but obscured this important aspect of ancient plant. Tobacco is considered to be an indigenous plant of [...] Read more →
The Apex Building, headquarters of the Federal Trade Commission, on Constitution Avenue and 7th Streets in Washington, D.C.. The building was designed by Edward H. Bennett under the purview of Secretary of the Treasury Andrew W. Mellon, and was completed in 1938 at a cost of $125 million. Photo by Carol M. Highsmith [...] Read more →
Note on Watercolour: F.A. Molony (fl. 1930-1938) was a Major in the Royal Engineers. The National Army Museum hold his work. His work was also shown at an exhibition of officers work at the R.B.A. Galleries (Army Officers’ Art Society) Description from Youtube: June 2015 will see [...] Read more →
Hunters at Work This is a recipe I created from scratch by trial and error. (Note: This recipe contains no eggs, refined white flour or white sugar.) 2 Cups Whole Wheat Flour – As unprocessed as you can find it 3 Cups of Raw Oatmeal 1 Cup of [...] Read more →
Cabildo circa 1936 The Cabildo houses a rare copy of Audubon’s Bird’s of America, a book now valued at $10 million+. Should one desire to visit the Cabildo, click here to gain free entry with a lowcost New Orleans Pass. Home Top of [...] Read more →
Home Top of Pg. Read more →
Quite possibly, the most agonizing decision being made by Baby Boomers across the nation these days is what to do with all that vintage Hi-fi equipment and boxes full of classic rock and roll cassettes and 8-Tracks. I faced this dilemma head-on this past summer as I definitely wanted in [...] Read more →
Toxicity of Rhododendron From Countrysideinfo.co.UK “Potentially toxic chemicals, particularly ‘free’ phenols, and diterpenes, occur in significant quantities in the tissues of plants of Rhododendron species. Diterpenes, known as grayanotoxins, occur in the leaves, flowers and nectar of Rhododendrons. These differ from species to species. Not all species produce them, although Rhododendron ponticum [...] Read more →
Filed under Miscellaneous. The Jubbulpore School of Industry is so thriving that the pupils, 800 in number, are obliged to work till ten o’clock at night to complete their orders; this they do most cheerfully. They are all Thugs, or the children of Thugs, and the hands which now ply [...] Read more →
VITRUVIUS The Ten Books on Architecture TRANSLATED By MORRIS HICKY MORGAN, PH.D., LL.D. LATE PROFESSOR OF CLASSICAL PHILOLOGY IN HARVARD UNIVERSITY WITH ILLUSTRATIONS AND ORIGINAL DESINGS PREPARED UNDER THE DIRECTION OF HERBERT LANGFORD WARREN, A.M. NELSON ROBINSON JR. PROFESSOR OF ARCHITECTURE IN HARVARD [...] Read more →
BLACKBERRY WINE 5 gallons of blackberries 5 pound bag of sugar Fill a pair of empty five gallon buckets half way with hot soapy water and a ¼ cup of vinegar. Wash thoroughly and rinse. Fill one bucket with two and one half gallons of blackberries and crush with [...] Read more →
Over the years I have observed a decline in manners amongst young men as a general principle and though there is not one particular thing that may be asserted as the causal reason for this, one might speculate… Self-awareness and being aware of one’s surroundings in social interactions [...] Read more →
Kenilworth Abbey Fields – Photo by David Hunt Click here to read Kenilworth by Sir Walter Scott Click here to view Kenilworth Notes Home Top of Pg. Read more →
Full Cover, rear, spine, and front Published by Piranesi Press in collaboration with Country House Essays, this beautiful paperback version of the King James Bible is now available for $79.95 at Barnes and Noble.com This is a limited Edition of 500 copies Worldwide. Click here to view other classic books [...] Read more →
Life insurance certificate issued by the Yorkshire Fire & Life Insurance Company to Samuel Holt, Liverpool, England, 1851. On display at the British Museum in London. Donated by the ifs School of Finance. Photo by Osama Shukir Muhammed Amin FRCP(Glasg) From How to Make Money; and How to Keep it, Or, Capital and Labor [...] Read more →
Mortlake Tapestries at Chatsworth House Click here to learn more about the Mortlake Tapestries of Chatsworth The Mortlake Tapestries were founded by Sir Francis Crane. From the Dictionary of National Biography, 1885-1900, Volume 13 Crane, Francis by William Prideaux Courtney CRANE, Sir FRANCIS (d. [...] Read more →
From Fores’s Sporting Notes and Sketches, A Quarterly Magazine Descriptive of British, Indian, Colonial, and Foreign Sport with Thirty Two Full Page Illustrations Volume 10 1893, London; Mssrs. Fores Piccadilly W. 1893, All Rights Reserved. GLIMPSES OF THE CHASE, Ireland a Hundred Years Ago. By ‘Triviator.’ FOX-HUNTING has, like Racing, [...] Read more →
Are you considering purchasing a copper water pitcher for storing drinking water but have questions about the effects on your health? The following study may help jump-start your research. Storing Drinking-water in Copper pots Kills Contaminating Diarrhoeagenic Bacteria ABSTRACT Microbially-unsafe water is [...] Read more →
http://www.vermeerscamera.co.uk/home.htm Home Top of Pg. Read more →
July, 16, l898 Forest and Stream Pg. 48 Tuna and Tarpon. New York, July 1.—Editor Forest and Stream: If any angler still denies the justice of my claim, as made in my article in your issue of July 2, that “the tuna is the grandest game [...] Read more →
ON THE ORIGIN OF SPECIES BY MEANS OF NATURAL SELECTION, OR THE PRESERVATION OF FAVOURED RACES IN THE STRUGGLE FOR LIFE. BY CHARLES DARWIN, M.A., FELLOW OF THE ROYAL, GEOLOGICAL, LINNÆAN, ETC., SOCIETIES ; AUTHOR OF ‘JOURNAL OF RESEARCHES DURING H.M.S. BEAGLE’S [...] Read more →
Home Top of Pg. Read more →
Audubon started to develop a special technique for drawing birds in 1806 a Mill Grove, Pennsylvania. He perfected it during the long river trip from Cincinnati to New Orleans and in New Orleans, 1821. Home Top of [...] Read more →
Sennen Cove at Dusk – photo by Jim Champion – 2005 Home Top of Pg. Read more →
Royal Exchange and The Bank of England From How to Make Money; and How to Keep it, Or, Capital and Labor based on the works of Thomas A. Davies Revised & Rewritten with Additions by Henry A. Ford A.M. – 1884 CHAPTER XXVI BANKING AND INSURANCE. I [...] Read more →
Oh Glorious England, verdant fields and wandering canals… In this wonderful series of videos, the CountryHouseGent takes the viewer along as he chugs up and down the many canals crisscrossing England in his classic Narrowboat. There is nothing like a free man charting his own destiny. Read more →
Home Top of [...] Read more →
If ever it could be said that there is such a thing as miracle healing soil, Ivan Sanderson said it best in his 1965 book entitled Ivan Sanderson’s Book of Great Jungles. Sanderson grew up with a natural inclination towards adventure and learning. He hailed from Scotland but spent much [...] Read more →
Reprint from The Royal Collection Trust website: Kneller was born in Lubeck, studied with Rembrandt in Amsterdam and by 1676 was working in England as a fashionable portrait painter. He painted seven British monarchs (Charles II, James II, William III, Mary II, Anne, George I and George II), though his [...] Read more →
Home Top of Pg. Read more →
Traditional British Christmas Pudding Recipe by Pen Vogler from the Charles Dickens Museum Ingredients 85 grams all purpose flour pinch of salt 170 grams Beef Suet 140 grams brown sugar tsp. mixed spice, allspice, cinnamon, cloves, &c 170 grams bread crumbs 170 grams raisins 170 grams currants 55 grams cut mixed peel Gram to [...] Read more →
Home Top of Pg. Read more →
Fred Kummerow on statin drugs (excerpt) from Jeremy Stuart on Vimeo. Dr. Kummerow passed away at the ripe old age of 102 in 2017. Click here to visit Dr. Mercola’s website. Home Top of Pg. Read more →
Aug. 13, 1898 Forest and Stream, Pg. 125 Game Bag and Gun. Indian Modes of Hunting. III.—Foxes. The fox as a rule is a most wily animal, and numerous are the stories of his cunning toward the Indian hunter with his steel traps. Read more →
July 2, 1898 Forest and Stream, Fresh-Water Angling. No. IX.—The Two Crappies. BY FRED MATHER. Fishing In Tree Tops. Here a short rod, say 8ft., is long enough, and the line should not be much longer than the rod. A reel is not [...] Read more →
Gate of Honour, Caius Court, Gonville & Caius Gonville & Caius College, known as Caius and pronounced keys was founded in 1348 by Edmund Gonville, the Rector of Terrington St Clement in Norfolk. The first name was thus Goville Hall and it was dedicated to the Annunciation of the Blessed Virgin Mary. [...] Read more →
It is unnecessary to point out that low-grade fruit may often be used to advantage in the preparation of vinegar. This has always been true in the case of apples and may be true with other fruit, especially grapes. The use of grapes for wine making is an outlet which [...] Read more →
Reprint from The Pitfalls of Speculation by Thomas Gibson 1906 Ed. THE PUBLIC ATTITUDE TOWARD SPECULATION THE public attitude toward speculation is generally hostile. Even those who venture frequently are prone to speak discouragingly of speculative possibilities, and to point warningly to the fact that an overwhelming majority [...] Read more →
Dried Norwegian Salt Cod Fried fish cakes are sold rather widely in delicatessens and at prepared food counters of department stores in the Atlantic coastal area. This product has possibilities for other sections of the country. Ingredients: Home Top of [...] Read more →
Citrus Fruit Culture THE PRINCIPAL fruit and nut trees grown commercially in the United States (except figs, tung, and filberts) are grown as varieties or clonal lines propagated on rootstocks. Almost all the rootstocks are grown from seed. The resulting seedlings then are either budded or grafted with propagating wood [...] Read more →
Country House Essays has returned after a good long summer holiday. More essays soon. Home Top of Pg. Read more →
San Felipe Model Reprinted from FineModelShips.com with the kind permission of Dr. Michael Czytko The SAN FELIPE is one of the most favoured ships among the ship model builders. The model is elegant, very beautifully designed, and makes a decorative piece of art to be displayed at home or in the [...] Read more →
Reprint from The Sportsman’s Cabinet and Town and Country Magazine, Vol I. Dec. 1832, Pg. 94-95 To the Editor of the Cabinet. SIR, Possessing that anxious feeling so common among shooters on the near approach of the 12th of August, I honestly confess I was not able [...] Read more →
The Queen Elizabeth Trust, or QEST, is an organisation dedicated to the promotion of British craftsmanship through the funding of scholarships and educational endeavours to include apprenticeships, trade schools, and traditional university classwork. The work of QEST is instrumental in keeping alive age old arts and crafts such as masonry, glassblowing, shoemaking, [...] Read more →
Home Top of Pg. Read more →
Donate to the YouTube site owner Gabe and he might send you some chocolate…. Home Top of Pg. Read more →
German made shotguns by Krieghoff, founded in 1886. Home Top of Pg. Read more →
Any prudent investor would jump at the chance to receive a guaranteed 6% dividend for life. So how does one get in on this action? The fact of the matter is…YOU can’t…That is unless you are a shareholder of one of the twelve Federal Reserve Banks and the banks under [...] Read more →
Wine Making Grapes are the world’s leading fruit crop and the eighth most important food crop in the world, exceeded only by the principal cereals and starchytubers. Though substantial quantities are used for fresh fruit, raisins, juice and preserves, most of the world’s annual production of about 60 million [...] Read more →
Home Top of Pg. Read more →
Dec. 10, 1898 Forest and Stream Pg. 477-479 Zulu. The little ship shown in the accompanying plans needs no description, as she speaks for herself, a handsome and shipshape craft that a man may own for years without any fear that she will go to pieces [...] Read more →
Home Top of Pg. Read more →
Biograph Theater, where John Dillinger was gunned down by the FBI on July 22, 1934 The Great Depression was on—highway based crime was rampant, the gangsters dressed as well as the bankers they robbed, and and Henry Ford’s big beautiful V8 sedan was the getaway car of choice for both wheelman and [...] Read more →
A terrestial globe on which the tracts and discoveries are laid down from the accurate observations made by Capts Cook, Furneux, Phipps, published 1782 / globe by John Newton ; cartography by William Palmer, held by the State Library of New South Wales The British Library, using sophisticated filming equipment and software, [...] Read more →
Home Top of Pg. Read more →
Home Top of Pg. Read more →
Crewe Hall Dining Room THE transient tenure that most of us have in our dwellings, and the absorbing nature of the struggle that most of us have to make to win the necessary provisions of life, prevent our encouraging the manufacture of well-wrought furniture. We mean to outgrow [...] Read more →
Felix Weihs de Weldon, age 96, died broke in the year 2003 after successive bankruptcies and accumulating $4 million dollars worth of debt. Most of the debt was related to the high cost of love for a wife living with Alzheimer’s. Health care costs to maintain his first wife, Margot, ran $500 per [...] Read more →
If a Woman asks you to Change, Politely Excuse Yourself and Walk out the Door; Forever Nobody changes; character is built early in life, and by the time one is involved in adult relationships, it is highly unlikely that one can rebuild one’s character. Recognizing this early on in ones adult [...] Read more →
The low level of work stoppages of recent years also attests to concern about job security. Testimony of Chairman Alan Greenspan The Federal Reserve’s semiannual monetary policy report Before the Committee on Banking, Housing, and Urban Affairs, U.S. Senate February 26, 1997 Iappreciate the opportunity to appear before this Committee [...] Read more →
Today I shall share a bit of market wisdom that will be hard to swallow for some Rolex owners, especially if they bought their Submariner at the top of the magical Covid Watch Bubble that has now collapsed. History often repeats itself, even in the stock market, but when [...] Read more →
Click here to view more great Italian recipes. Home Top of Pg. Read more →
Book Conservators, Mitchell Building, State Library of New South Wales, 29.10.1943, Pix Magazine The following is taken verbatim from a document that appeared several years ago in the Maine State Archives. It seems to have been removed from their website. I happened to have made a physical copy of it at the [...] Read more →
Laurens’ portrait as painted during his time spent imprisoned in the Tower of London, where he was kept for over a year after being captured at sea while serving as the United States minister to the Netherlands during the Revolutionary War. The first Christian white man to be cremated in America was [...] Read more →
Home Top of Pg. Read more →
July 9, 1898. Forest and Stream Pg. 25 Some Notes on American Ship-Worms. [Read before the American Fishes Congress at Tampa.] While we wish to preserve and protect most of the products of our waters, these creatures we would gladly obliterate from the realm of living things. For [...] Read more →
Click here to read the Swiss National Bank’s Chronicle of Monetary Events. Home Top of Pg. Read more →
STORE MANAGEMENT—THE SHIRK. THE shirk is a well-known specimen of the genus homo. His habitat is offices, stores, business establishments of all kinds. His habits are familiar to us, but a few words on the subject will not be amiss. The shirk usually displays activity when the boss is around, [...] Read more →
Welcome to Country House Essays. Enjoy. Home Top of Pg. Read more →
Home Top of Pg. Read more →
Bess of Harwick Four times the nuptial bed she warm’d, And every time so well perform’d, That when death spoil’d each husband’s billing, He left the widow every shilling. Fond was the dame, but not dejected; Five stately mansions she erected With more than royal pomp, to vary The prison of her captive When [...] Read more →
PAINTER-WORK, in the building trade. When work is painted one or both of two distinct ends is achieved, namely the preservation and the coloration of the material painted. The compounds used for painting—taking the word as meaning a thin protective or decorative coat—are very numerous, including oil-paint of many kinds, distemper, whitewash, [...] Read more →
Blackbeard’s Jolly Roger If you’re looking for that most refreshing of summertime beverages for sipping out on the back patio or perhaps as a last drink before walking the plank, let me recommend my Blunderbuss Mai Tai. I picked up the basics to this recipe over thirty years ago when holed up [...] Read more →
St.Helen’s on the Thames, photo by Momit From a Dictionary of the Thames from Oxford to the Nore. 1880 by Charles Dickens Abingdon, Berkshire, on the right bank, from London 103 3/4miles, from Oxford 7 3/4 miles. A station on the Great Western Railway, from Paddington 60 miles. The time occupied [...] Read more →
WITCHCRAFT, SORCERY, MAGIC AND OTHER PSYCHOLOGICAL PHENOMENA AND THEIR IMPLICATIONS ON MILITARY AND PARAMILITARY OPERATIONS IN THE CONGO This report has been prepared in response to a query posed by ODCS/OPS, Department of the Army, regarding the purported use of witchcraft, sorcery, and magic by insurgent elements in the Republic [...] Read more →
AB Bookman’s 1948 Guide to Describing Conditions: As New is self-explanatory. It means that the book is in the state that it should have been in when it left the publisher. This is the equivalent of Mint condition in numismatics. Fine (F or FN) is As New but allowing for the normal effects of [...] Read more →
Home Top of Pg. Read more →
The following recipes are from a small booklet entitled 500 Delicious Salads that was published for the Culinary Arts Institute in 1940 by Consolidated Book Publishers, Inc. 153 N. Michigan Ave., Chicago, Ill. If you have been looking for a way to lighten up your salads and be free of [...] Read more →
From The How and When, An Authoritative reference reference guide to the origin, use and classification of the world’s choicest vintages and spirits by Hyman Gale and Gerald F. Marco. The Marco name is of a Chicago family that were involved in all aspects of the liquor business and ran Marco’s Bar [...] Read more →
A rhetorical question? Genuine concern? In this essay we are examining another form of matter otherwise known as national literary matters, the three most important of which being the Matter of Rome, Matter of France, and the Matter of England. Our focus shall be on the Matter of England or [...] Read more →
Home Top of Pg. Read more →
Hebborn Piranesi Before meeting with an untimely death at the hand of an unknown assassin in Rome on January 11th, 1996, master forger Eric Hebborn put down on paper a wealth of knowledge about the art of forgery. In a book published posthumously in 1997, titled The Art Forger’s Handbook, Hebborn suggests [...] Read more →
Muscadine Jelly 6 cups muscadine grape juice 6 cups sugar 1 box Kraft Sure Gel or Ball Fruit Jell Home Top of [...] Read more →
EBAY’S FRAUD PROBLEM IS GETTING WORSE EBay has had a problem with fraudulent sellers since its inception back in 1995. Some aspects of the platform have improved with algorithms and automation, but others such as customer service and fraud have gotten worse. Small sellers have definitely been hurt by eBay’s [...] Read more →
Click here to access the world’s most powerful Import/Export Research Database on the Planet. With this search engine one is able to access U.S. Customs and other government data showing suppliers for any type of company in the United States. Home Top of Pg. Read more →
Home Top of Pg. Read more →
Video courtesy of Imperial War Museums, UK Home Top of Pg. Read more →
Vintage woodcut illustration of a Eel This dish is a favorite in Northern Europe, from the British Isles to Sweden. Clean and skin the eels and cut them into pieces about 3/4-inch thick. Wash and drain the pieces, then dredge in fine salt and allow to stand from 30 [...] Read more →
NAPOLEON’S PHARMACISTS. Of the making of books about Napoleon there is no end, and the centenary of his death (May 5) is not likely to pass without adding to the number, but a volume on Napoleon”s pharmacists still awaits treatment by the student in this field of historical research. There [...] Read more →
A Lecture Delivered at the Guildhall, March 2, 1853 by Rev. H.M. Scarth, M.A., Rector of Bathwick. To understand the ancient history of the country in which we live, to know something of the arts and manners of the people who have preceded us, to ascertain what we owe [...] Read more →
The following are transcripts of two letters written by the Founding Father Thomas Jefferson on the subject of seed saving. “November 27, 1818. Monticello. Thomas Jefferson to Henry E. Watkins, transmitting succory seed and outlining the culture of succory.” [Transcript] Thomas Jefferson Correspondence Collection Collection 89 Read more →
The existence of large bodies of men having no other means of subsistence than those afforded by plunder, is, in all countries, too common to excite surprise; and, unhappily, organized bands of assassins are not peculiar to India! The associations of murderers known by the name of Thugs present, however, [...] Read more →
Cleaner for Gilt Frames. Calcium hypochlorite…………..7 oz. Sodium bicarbonate……………7 oz. Sodium chloride………………. 2 oz. Distilled water…………………12 oz. Home Top of Pg. Read more →
1 garlic clove, cut in half 1 8-oz. pkg. Philadelphia Brand Cream Cheese 3 tablespoons clam broth 1 7-to 7 1/2-oz. can minced clams, drained Home Top of [...] Read more →
Mocking Bird Food. Hemp seed……….2 pounds Rape seed………. .1 pound Crackers………….1 pound Rice…………….1/4 pound Corn meal………1/4 pound Lard oil…………1/4 pound Home Top of Pg. Read more →
|
Charles Dickens wrote much more than novels. In fact he turned out several very interesting dictionaries to include one of London, one of Paris and one on London’s long meandering river Thames. Click here to read a copy of the Dictionary of the Thames. Home Top of Pg. Read more →
The Racing Knockabout Gosling. Gosling was the winning yacht of 1897 in one of the best racing classes now existing in this country, the Roston knockabout class. The origin of this class dates back about six years, when Carl, a small keel cutter, was built for C. H. [...] Read more →
EBAY’S FRAUD PROBLEM IS GETTING WORSE EBay has had a problem with fraudulent sellers since its inception back in 1995. Some aspects of the platform have improved with algorithms and automation, but others such as customer service and fraud have gotten worse. Small sellers have definitely been hurt by eBay’s [...] Read more →
Click here to visit Lil’ Lost Lou and purchase a copy of her latest album. Home Top of Pg. Read more →
Blackbeard’s Jolly Roger If you’re looking for that most refreshing of summertime beverages for sipping out on the back patio or perhaps as a last drink before walking the plank, let me recommend my Blunderbuss Mai Tai. I picked up the basics to this recipe over thirty years ago when holed up [...] Read more →
Home Top of [...] Read more →
Any prudent investor would jump at the chance to receive a guaranteed 6% dividend for life. So how does one get in on this action? The fact of the matter is…YOU can’t…That is unless you are a shareholder of one of the twelve Federal Reserve Banks and the banks under [...] Read more →
The existence of large bodies of men having no other means of subsistence than those afforded by plunder, is, in all countries, too common to excite surprise; and, unhappily, organized bands of assassins are not peculiar to India! The associations of murderers known by the name of Thugs present, however, [...] Read more →
Dr. David Starkey, the UK’s premiere historian, speaks to the modern and fleeting notion of “cancel culture”. Starkey’s brilliance is unparalleled and it has become quite obvious to the world’s remaining Western scholars willing to stand on intellectual integrity that a few so-called “Woke Intellectuals” most certainly cannot undermine [...] Read more →
July 2, 1898 Forest and Stream, Fresh-Water Angling. No. IX.—The Two Crappies. BY FRED MATHER. Fishing In Tree Tops. Here a short rod, say 8ft., is long enough, and the line should not be much longer than the rod. A reel is not [...] Read more →
WIPO HQ Geneva UNITED STATES PLANT VARIETY PROTECTION ACT TITLE I – PLANT VARIETY PROTECTION OFFICE Chapter Section 1. Organization and Publications . 1 2. Legal Provisions as to the Plant Variety Protection Office . 21 3. Plant Variety Protection Fees . 31 CHAPTER 1.-ORGANIZATION AND PUBLICATIONS Section [...] Read more →
Donate to the YouTube site owner Gabe and he might send you some chocolate…. Home Top of Pg. Read more →
Testing the Irish Blue Terrier Breed in 1923. Home Top of Pg. Read more →
Jul. 23, 1898 Forest and Stream, Pg. 65 Horn Measurements. Editor Forest and Stream: “Record head.” How shamefully this term is being abused, especially in the past three years; or since the giant moose from Alaska made his appearance in public and placed all former records (so far as [...] Read more →
INTRODUCTION The idea of compiling this little volume occurred to me while on a visit to some friends at their summer home in a quaint New England village. The little town had once been a thriving seaport, but now consisted of hardly more than a dozen old-fashioned Colonial houses facing [...] Read more →
Cleaner for Gilt Frames. Calcium hypochlorite…………..7 oz. Sodium bicarbonate……………7 oz. Sodium chloride………………. 2 oz. Distilled water…………………12 oz. Home Top of Pg. Read more →
FRIED SQUIRREL & BISCUIT GRAVY 3-4 Young Squirrels, dressed and cleaned 1 tsp. Morton Salt or to taste 1 tsp. McCormick Black Pepper or to taste 1 Cup Martha White All Purpose Flour 1 Cup Hog Lard – Preferably fresh from hog killing, or barbecue table Cut up three to [...] Read more →
Home Top of Pg. Read more →
THE answer to the question, What is fortune has never been, and probably never will be, satisfactorily made. What may be a fortune for one bears but small proportion to the colossal possessions of another. The scores or hundreds of thousands admired and envied as a fortune in most of our communities [...] Read more →
PAINTER-WORK, in the building trade. When work is painted one or both of two distinct ends is achieved, namely the preservation and the coloration of the material painted. The compounds used for painting—taking the word as meaning a thin protective or decorative coat—are very numerous, including oil-paint of many kinds, distemper, whitewash, [...] Read more →
Bess of Harwick Four times the nuptial bed she warm’d, And every time so well perform’d, That when death spoil’d each husband’s billing, He left the widow every shilling. Fond was the dame, but not dejected; Five stately mansions she erected With more than royal pomp, to vary The prison of her captive When [...] Read more →
Welcome to Country House Essays. Enjoy. Home Top of Pg. Read more →
Aw, the good old days, meet in the coffee shop with a few friends, click open the Zippo, inhale a glorious nosegay of lighter fluid, fresh roasted coffee and a Marlboro cigarette…. A Meta-analysis of Coffee Drinking, Cigarette Smoking, and the Risk of Parkinson’s Disease We conducted a [...] Read more →
Chen Lin, Water fowl, in Cahill, James. Ge jiang shan se (Hills Beyond a River: Chinese Painting of the Yuan Dynasty, 1279-1368, Taiwan edition). Taipei: Shitou chubanshe fen youxian gongsi, 1994. pl. 4:13, p. 180. Collection of the National Palace Museum, Taipei. scroll, light colors on paper, 35.7 x 47.5 cm Read more →
What is follows is an historical article that appeared in The Hartford Courant in 1916 about the arsenic murders carried out by Mrs. Archer-Gilligan. This story is the basis for the 1944 Hollywood film “Arsenic and Old Lace” starring Cary Grant and Priscilla Lane and directed by Frank Capra. The [...] Read more →
Home Top of Pg. Read more →
Home Top of Pg. Read more →
Artisans world-wide spend a fortune on commercial brand oil-based gold leaf sizing. The most popular brands include Luco, Dux, and L.A. Gold Leaf. Pricing for quart size containers range from $35 to $55 depending upon retailer pricing. Fast drying sizing sets up in 2-4 hours depending upon environmental conditions, humidity [...] Read more →
Add the following ingredients to a four or six quart crock pot, salt & pepper to taste keeping in mind that salt pork is just that, cover with water and cook on high till it boils, then cut back to low for four or five hours. A slow cooker works well, I [...] Read more →
Reprint from The Pitfalls of Speculation by Thomas Gibson 1906 Ed. THE PUBLIC ATTITUDE TOWARD SPECULATION THE public attitude toward speculation is generally hostile. Even those who venture frequently are prone to speak discouragingly of speculative possibilities, and to point warningly to the fact that an overwhelming majority [...] Read more →
Home Top of Pg. Read more →
Vintage woodcut illustration of a Eel This dish is a favorite in Northern Europe, from the British Isles to Sweden. Clean and skin the eels and cut them into pieces about 3/4-inch thick. Wash and drain the pieces, then dredge in fine salt and allow to stand from 30 [...] Read more →
From Allen’s Indian Mail, December 3rd, 1851 BOMBAY. MUSULMAN FANATICISM. On the evening of November 15th, the little village of Mahim was the scene of a murder, perhaps the most determined which has ever stained the annals of Bombay. Three men were massacred in cold blood, in a house used [...] Read more →
An angler with a costly pole Surmounted with a silver reel, Carven in quaint poetic scroll- Jointed and tipped with finest steel— With yellow flies, Whose scarlet eyes And jasper wings are fair to see, Hies to the stream Whose bubbles beam Down murmuring eddies wild and free. And casts the line with sportsman’s [...] Read more →
Buying a book for a serious collector with refined tastes can be a daunting task. However, there is one company that publishes some of the finest reproduction books in the world, books that most collectors wouldn’t mind having in their collection no matter their general preference or specialty. Read more →
Muscadine Jelly 6 cups muscadine grape juice 6 cups sugar 1 box Kraft Sure Gel or Ball Fruit Jell Home Top of [...] Read more →
Home Top of Pg. Read more →
WITCHCRAFT, SORCERY, MAGIC AND OTHER PSYCHOLOGICAL PHENOMENA AND THEIR IMPLICATIONS ON MILITARY AND PARAMILITARY OPERATIONS IN THE CONGO This report has been prepared in response to a query posed by ODCS/OPS, Department of the Army, regarding the purported use of witchcraft, sorcery, and magic by insurgent elements in the Republic [...] Read more →
The Hunt Saboteur is a national disgrace barking out loud, black mask on her face get those dogs off, get them off she did yell until a swift kick from me mare her voice it did quell and sent the Hunt Saboteur scurrying up vale to the full cry of hounds drowning out her [...] Read more →
The element copper effectively kills viruses and bacteria. Therefore it would reason and I will assert and not only assert but lay claim to the patents for copper mesh stints to be inserted in the arteries of patients presenting with severe cases of Covid-19 with a slow release dosage of [...] Read more →
Royal Exchange and The Bank of England From How to Make Money; and How to Keep it, Or, Capital and Labor based on the works of Thomas A. Davies Revised & Rewritten with Additions by Henry A. Ford A.M. – 1884 CHAPTER XXVI BANKING AND INSURANCE. I [...] Read more →
KING ARTHUR AND HIS KNIGHTS On the decline of the Roman power, about five centuries after Christ, the countries of Northern Europe were left almost destitute of a national government. Numerous chiefs, more or less powerful, held local sway, as far as each could enforce his dominion, and occasionally those [...] Read more →
Reprint from the Sportsman Cabinet and Town & Country Magazine, Vol.1, Number 1, November 1832. MR. Editor, Will you allow me to inquire, through the medium of your pages, the correct meaning of the term thorough-bred fox-hound? I am very well aware, that the expression is in common [...] Read more →
Como dome facade – Pliny the Elder – Photo by Wolfgang Sauber Work in Progress… THE VARNISHES. Every substance may be considered as a varnish, which, when applied to the surface of a solid body, gives it a permanent lustre. Drying oil, thickened by exposure to the sun’s heat or [...] Read more →
From the classic British Movie, The Shooting Party, a 1985 British drama film directed by Alan Bridges based on Isabel Colegate’s 9th novel of the same name published in 1980 we find a scene set in the billiards parlor whereupon the host of the weekend shooting party Sir Randolph Nettleby walks in [...] Read more →
St.Helen’s on the Thames, photo by Momit From a Dictionary of the Thames from Oxford to the Nore. 1880 by Charles Dickens Abingdon, Berkshire, on the right bank, from London 103 3/4miles, from Oxford 7 3/4 miles. A station on the Great Western Railway, from Paddington 60 miles. The time occupied [...] Read more →
Full Cover, rear, spine, and front Published by Piranesi Press in collaboration with Country House Essays, this beautiful paperback version of the King James Bible is now available for $79.95 at Barnes and Noble.com This is a limited Edition of 500 copies Worldwide. Click here to view other classic books [...] Read more →
Filed under Miscellaneous. The Jubbulpore School of Industry is so thriving that the pupils, 800 in number, are obliged to work till ten o’clock at night to complete their orders; this they do most cheerfully. They are all Thugs, or the children of Thugs, and the hands which now ply [...] Read more →
Salmon and Sturgeon Caviar – Photo by Thor Salmon caviar was originated about 1910 by a fisherman in the Maritime Provinces of Siberia, and the preparation is a modification of the sturgeon caviar method (Cobb 1919). Salomon caviar has found a good market in the U.S.S.R. and other European countries where it [...] Read more →
To Choose Poultry. When fresh, the eyes should be clear and not sunken, the feet limp and pliable, stiff dry feet being a sure indication that the bird has not been recently killed; the flesh should be firm and thick and if the bird is plucked there should be no [...] Read more →
BOOKS CONDEMNED TO BE BURNT. By JAMES ANSON FARRER, LONDON ELLIOT STOCK, 62, PATERNOSTER ROW 1892 ———- WHEN did books first come to be burnt in England by the common hangman, and what was [...] Read more →
To Clean Watch Chains. Gold or silver watch chains can be cleaned with a very excellent result, no matter whether they may be matt or polished, by laying them for a few seconds in pure aqua ammonia; they are then rinsed in alcohol, and finally. shaken in clean sawdust, free from sand. [...] Read more →
The Effect of Magnetic Fields on Wound Healing Experimental Study and Review of the Literature Steven L. Henry, MD, Matthew J. Concannon, MD, and Gloria J. Yee, MD Division of Plastic Surgery, University of Missouri Hospital & Clinics, Columbia, MO Published July 25, 2008 Objective: Magnets [...] Read more →
Home Top of Pg. Read more →
Home Top of Pg. Read more →
Home Top of Pg. Read more →
King George IV was known far and wide as the dandy king, incompetent, ugly, and vulgar. As Prince regent, prior to his assent to the throne, he kept fast company with Beau Brummel, King of Dandies, a man sixteen years his younger. And decadence followed. King George was a gambler, philanderer, and [...] Read more →
Silverfish damage to book – photo by Micha L. Rieser The beauty of hunting silverfish is that they are not the most clever of creatures in the insect kingdom. Simply take a small clean glass jar and wrap it in masking tape. The masking tape gives the silverfish something to [...] Read more →
Home Top of Pg. Read more →
Home Top of Pg. Read more →
Reprint from The Royal Collection Trust website: Kneller was born in Lubeck, studied with Rembrandt in Amsterdam and by 1676 was working in England as a fashionable portrait painter. He painted seven British monarchs (Charles II, James II, William III, Mary II, Anne, George I and George II), though his [...] Read more →
Crewe Hall Dining Room THE transient tenure that most of us have in our dwellings, and the absorbing nature of the struggle that most of us have to make to win the necessary provisions of life, prevent our encouraging the manufacture of well-wrought furniture. We mean to outgrow [...] Read more →
William Wyggeston’s chantry house, built around 1511, in Leicester: The building housed two priests, who served at a chantry chapel in the nearby St Mary de Castro church. It was sold as a private dwelling after the dissolution of the chantries. A Privately Built Chapel Chantry, chapel, generally within [...] Read more →
Click here to read the Swiss National Bank’s Chronicle of Monetary Events. Home Top of Pg. Read more →
Home Top of Pg. Read more →
The Ardabil Carpet – Made in the town of Ardabil in north-west Iran, the burial place of Shaykh Safi al-Din Ardabili, who died in 1334. The Shaykh was a Sufi leader, ancestor of Shah Ismail, founder of the Safavid dynasty (1501-1722). While the exact origins of the carpet are unclear, it’s believed to have [...] Read more →
THE FOWLING PIECE, from the Shooter’s Guide by B. Thomas – 1811. I AM perfectly aware that a large volume might be written on this subject; but, as my intention is to give only such information and instruction as is necessary for the sportsman, I shall forbear introducing any extraneous [...] Read more →
Click here to read the Condon Report Home Top of Pg. Read more →
Quite possibly, the most agonizing decision being made by Baby Boomers across the nation these days is what to do with all that vintage Hi-fi equipment and boxes full of classic rock and roll cassettes and 8-Tracks. I faced this dilemma head-on this past summer as I definitely wanted in [...] Read more →
Hudson Bay: Trappers, 1892. N’Talking Musquash.’ Fur Trappers Of The Hudson’S Bay Company Talking By A Fire. Engraving After A Drawing By Frederic Remington, 1892. Indian Modes of Hunting. IV.—Musquash. In Canada and the United States, the killing of the little animal known under the several names of [...] Read more →
Home Top of Pg. Read more →
Dutch artist Herman de Vries – Photo taken by son Vince The two videos below of Herman de Vries at work at the Venice Bienalle 2015 are quite inspiring. So inspiring in fact that I moved into a cave for two weeks and wrote Shakespearean tragedy with charcoal. Filled with great joy [...] Read more →
Gate of Honour, Caius Court, Gonville & Caius Gonville & Caius College, known as Caius and pronounced keys was founded in 1348 by Edmund Gonville, the Rector of Terrington St Clement in Norfolk. The first name was thus Goville Hall and it was dedicated to the Annunciation of the Blessed Virgin Mary. [...] Read more →
It was a strange assignment. I picked up the telegram from desk and read it a third time. NEW YORK, N.Y., MAY 9, 1949 HAVE BEEN INVESTIGATING FLYING SAUCER MYSTERY. FIRST TIP HINTED GIGANTIC HOAX TO COVER UP OFFICIAL SECRET. BELIEVE IT MAY HAVE BEEN PLANTED TO HIDE [...] Read more →
Foie gras with Sauternes, Photo by Laurent Espitallier As an Appetizer Pale dry Sherry, with or without bitters, chilled or not. Plain or mixed Vermouth, with or without bitters. A dry cocktail. With Oysters, Clams or Caviar A dry flinty wine such as Chablis, Moselle, Champagne. Home Top of [...] Read more →
From The How and When, An Authoritative reference reference guide to the origin, use and classification of the world’s choicest vintages and spirits by Hyman Gale and Gerald F. Marco. The Marco name is of a Chicago family that were involved in all aspects of the liquor business and ran Marco’s Bar [...] Read more →
New York Stock Exchange Floor September 26,1963 The Specialist as a member of a stock exchange has two functions.’ He must execute orders which other members of an exchange may leave with him when the current market price is away from the price of the orders. By executing these orders on behalf [...] Read more →
by John Partridge,drawing,1825 From the work of Sir Charles Lock Eastlake entitled Materials for a history of oil painting, (London: Longman, Brown, Green, and Longmans, 1846), we learn the following: The effect of oil at certain temperatures, in penetrating “the minute pores of the amber” (as Hoffman elsewhere writes), is still more [...] Read more →
AB Bookman’s 1948 Guide to Describing Conditions: As New is self-explanatory. It means that the book is in the state that it should have been in when it left the publisher. This is the equivalent of Mint condition in numismatics. Fine (F or FN) is As New but allowing for the normal effects of [...] Read more →
Jan Verkolje Antonie van Leeuwenhoek was the first person to describe gout or uric acid crystals 1679. For one suffering gout, the following vitamins, herbs, and extracts may be worth looking into: Vitamin C Folic Acid – Folic Acid is a B vitamin and is also known as B9 – [Known food [...] Read more →
Mudlarks of London Mudlarking along the Thames River foreshore is controlled by the Port of London Authority. According to the Port of London website, two type of permits are issued for those wishing to conduct metal detecting, digging, or searching activities. Standard – allows digging to a depth of 7.5 [...] Read more →
Patek Phillipe hand makes the finest watches in the world. Click here to learn more. Home Top of Pg. Read more →
BEEF JERKY Preparation. Slice 5 pounds lean beef (flank steak or similar cut) into strips 1/8 to 1/4 inch thick, 1 to 2 inches wide, and 4 to 12 inches long. Cut with grain of meat; remove the fat. Lay out in a single layer on a smooth clean surface (use [...] Read more →
Home Top of Pg. Read more →
Cabildo circa 1936 The Cabildo houses a rare copy of Audubon’s Bird’s of America, a book now valued at $10 million+. Should one desire to visit the Cabildo, click here to gain free entry with a lowcost New Orleans Pass. Home Top of [...] Read more →
A Real Soda Jerk FORMULAS FROM VARIOUS SOURCES. Pineapple Frappe. Water, 1 gallon; sugar 2 pounds of water. 61/2 pints, and simple syrup. 2 1/2 pints; 2 pints of pineapple stock or 1 pint of pineapple stock and 1 pint of grated pineapple juice of 6 lemons. Mix, [...] Read more →
PEACH BRANDY 2 gallons + 3 quarts boiled water 3 qts. peaches, extremely ripe 3 lemons, cut into sections 2 sm. pkgs. yeast 10 lbs. sugar 4 lbs. dark raisins Place peaches, lemons and sugar in crock. Dissolve yeast in water (must NOT be to hot). Stir thoroughly. Stir daily for 7 days. Keep [...] Read more →
Home Top of Pg. Read more →
VHF Marifoon Sailor RT144, by S.J. de Waard RADIO INFORMATION FOR BOATERS Effective 01 August, 2013, the U. S. Coast Guard terminated its radio guard of the international voice distress, safety and calling frequency 2182 kHz and the international digital selective calling (DSC) distress and safety frequency 2187.5 kHz. Additionally, [...] Read more →
Baking is a very similar process to roasting: the two often do duty for one another. As in all other methods of cookery, the surrounding air may be several degrees hotter than boiling water, but the food is no appreciably hotter until it has lost water by evaporation, after which it may [...] Read more →
Dominion, Royal St. Lawrence Yacht Club,Winner of Seawanhaka Cup, 1898. The Tail Wags the Dog. The following is a characteristic sample of those broad and liberal views on yachting which are the pride of the Boston Herald. Speaking of the coming races for the Seawanhaka international challenge cup, it says: [...] Read more →
THE SNIPE, from the Shooter’s Guide by B. Thomas – 1811 AFTER having given a particular description of the woodcock, it will only. be necessary to observe, that the plumage and shape of the snipe is much the same ; and indeed its habits and manners sets bear a great [...] Read more →
” Here’s many a year to you ! Sportsmen who’ve ridden life straight. Here’s all good cheer to you ! Luck to you early and late. Here’s to the best of you ! You with the blood and the nerve. Here’s to the rest of you ! What of a weak moment’s swerve ? [...] Read more →
Photo by Rebecca Humann Texas Tea Recipe 2 oz Cuervo Gold Tequila Home Top of [...] Read more →
Gold Home Top of Pg. Read more →
San Felipe Model Reprinted from FineModelShips.com with the kind permission of Dr. Michael Czytko The SAN FELIPE is one of the most favoured ships among the ship model builders. The model is elegant, very beautifully designed, and makes a decorative piece of art to be displayed at home or in the [...] Read more →
A terrestial globe on which the tracts and discoveries are laid down from the accurate observations made by Capts Cook, Furneux, Phipps, published 1782 / globe by John Newton ; cartography by William Palmer, held by the State Library of New South Wales The British Library, using sophisticated filming equipment and software, [...] Read more →
Jujitsu training 1920 in Japanese agricultural school. CHAPTER V THE VALUE OF EVEN TEMPER IN ATHLETICS—SOME OF THE FEATS THAT REQUIRE GOOD NATURE In the writer’s opinion it becomes necessary to make at this point some suggestions relative to a very important part of the training in jiu-jitsu. [...] Read more →
From Dr. Marvel’s 1929 book entitled Hoodoo for the Common Man, we find his infamous Hoochie Coochie Hex. What follows is a verbatim transcription of the text: The Hoochie Coochie Hex should not be used in conjunction with any other Hexes. This can lead to [...] Read more →
Home Top of Pg. Read more →
A la Russie, aux ânes et aux autres – by Chagall – 1911 Marc Chagall is one of the most forged artists on the planet. Mark Rothko fakes also abound. According to available news reports, the art market is littered with forgeries of their work. Some are even thought to be [...] Read more →
Roger Scruton – Why Beauty Matters (2009) from Mirza Akdeniz on Vimeo. Click here for another site on which to view this video. Sadly, Sir Roger Scruton passed away a few days ago—January 12th, 2020. Heaven has gained a great philosopher. Home Top of [...] Read more →
Over the years I have observed a decline in manners amongst young men as a general principle and though there is not one particular thing that may be asserted as the causal reason for this, one might speculate… Self-awareness and being aware of one’s surroundings in social interactions [...] Read more →
Click here to view more great Italian recipes. Home Top of Pg. Read more →
Reprint from the Royal Collection Trust Website The meeting between Henry VIII and Francis I, known as the Field of the Cloth of Gold, took place between 7 to 24 June 1520 in a valley subsequently called the Val d’Or, near Guisnes to the south of Calais. The [...] Read more →
Home Top of Pg. Read more →
BLACKBERRY WINE 5 gallons of blackberries 5 pound bag of sugar Fill a pair of empty five gallon buckets half way with hot soapy water and a ¼ cup of vinegar. Wash thoroughly and rinse. Fill one bucket with two and one half gallons of blackberries and crush with [...] Read more →
Home Top of Pg. Read more →
TROF. C. F. HOLDFER AND HIS 183LBS. TUNA, WITH BOATMAN JIM GARDNER. July 2, 1898. Forest and Stream Pg. 11 The Tuna Record. Avalon. Santa Catalina Island. Southern California, June 16.—Editor Forest and Stream: Several years ago the writer in articles on the “Game Fishes of the Pacific Slope,” in [...] Read more →
Home Top of Pg. Read more →
Cleremont Club 44 Berkeley Square, London Home Top of Pg. Read more →
Click here to learn more about The Tallis Scholars Home Top of Pg. Read more →
Home Top of Pg. Read more →
Hebborn Piranesi Before meeting with an untimely death at the hand of an unknown assassin in Rome on January 11th, 1996, master forger Eric Hebborn put down on paper a wealth of knowledge about the art of forgery. In a book published posthumously in 1997, titled The Art Forger’s Handbook, Hebborn suggests [...] Read more →
Mortlake Tapestries at Chatsworth House Click here to learn more about the Mortlake Tapestries of Chatsworth The Mortlake Tapestries were founded by Sir Francis Crane. From the Dictionary of National Biography, 1885-1900, Volume 13 Crane, Francis by William Prideaux Courtney CRANE, Sir FRANCIS (d. [...] Read more →
Home Top of Pg. Read more →
NAPOLEON’S PHARMACISTS. Of the making of books about Napoleon there is no end, and the centenary of his death (May 5) is not likely to pass without adding to the number, but a volume on Napoleon”s pharmacists still awaits treatment by the student in this field of historical research. There [...] Read more →
Are you considering purchasing a copper water pitcher for storing drinking water but have questions about the effects on your health? The following study may help jump-start your research. Storing Drinking-water in Copper pots Kills Contaminating Diarrhoeagenic Bacteria ABSTRACT Microbially-unsafe water is [...] Read more →
Home Top of [...] Read more →
Sebastian Cox is one of the UK’s premier custom furniture makers with a unique background and love for the forest. Click here to visit SebastianCox.co.uk Home Top of Pg. Read more →
Home Top of Pg. Read more →
For years in the West African nation of Ghana medicine men have used a root and leaves from a plant called nibima(Cryptolepis sanguinolenta) to kill the Plasmodium parasite transmitted through a female mosquito’s bite that is the root cause of malaria. A thousand miles away in India, a similar(same) plant [...] Read more →
Home Top of Pg. Read more →
Hunters at Work This is a recipe I created from scratch by trial and error. (Note: This recipe contains no eggs, refined white flour or white sugar.) 2 Cups Whole Wheat Flour – As unprocessed as you can find it 3 Cups of Raw Oatmeal 1 Cup of [...] Read more →
Click here to read the full text of the Hunting Act – 2004 Home Top of Pg. Read more →
Banana Propagation Reprinted from the International Institute of Tropical Agriculture (IITA.org) The traditional means of obtaining banana planting material (“seed”) is to acquire suckers from one’s own banana garden, from a neighbor, or from a more distant source. This method served to spread common varieties around the world and to multiply them [...] Read more →
Photo Caption: The Marquis of Zetland, KC, PC – otherwise known as Lawrence Dundas Son of: John Charles Dundas and: Margaret Matilda Talbot born: Friday 16 August 1844 died: Monday 11 March 1929 at Aske Hall Occupation: M.P. for Richmond Viceroy of Ireland Vice Lord Lieutenant of North Yorkshire Lord – in – Waiting [...] Read more →
Home Top of Pg. Read more →
1 garlic clove, cut in half 1 8-oz. pkg. Philadelphia Brand Cream Cheese 3 tablespoons clam broth 1 7-to 7 1/2-oz. can minced clams, drained Home Top of [...] Read more →
Armorial tablet of the Stewarts – Falkland Palace Fife, Scotland. The Stewart Kings – King James I & VI to Charles II Six video playlist on the Kings of England: Home Top of Pg. Read more →
Home Top of Pg. Read more →
? Home Top of Pg. Read more →
If ever it could be said that there is such a thing as miracle healing soil, Ivan Sanderson said it best in his 1965 book entitled Ivan Sanderson’s Book of Great Jungles. Sanderson grew up with a natural inclination towards adventure and learning. He hailed from Scotland but spent much [...] Read more →
The 1896 Victorian terracotta Bell Edison Telephone Building – 17 & 19 Newhall Street, Birmingham, England. A grade I listed building designed by Frederick Martin of the firm Martin & Chamberlain. Now offices for firms of architects. Photographed 10 May 2006 by Oosoom [Reprint from Victoria and Albert Museum included below on [...] Read more →
THE HATHA YOGA PRADIPIKA Translated into English by PANCHAM SINH Panini Office, Allahabad [1914] INTRODUCTION. There exists at present a good deal of misconception with regard to the practices of the Haṭha Yoga. People easily believe in the stories told by those who themselves [...] Read more →
The following are transcripts of two letters written by the Founding Father Thomas Jefferson on the subject of seed saving. “November 27, 1818. Monticello. Thomas Jefferson to Henry E. Watkins, transmitting succory seed and outlining the culture of succory.” [Transcript] Thomas Jefferson Correspondence Collection Collection 89 Read more →
*note – Billesdon and Billesden have both been used to name the hunt. BILLESDEN COPLOW POEM [From “Reminiscences of the late Thomas Assheton Smith, Esq”] The run celebrated in the following verses took place on the 24th of February, 1800, when Mr. Meynell hunted Leicestershire, and has since been [...] Read more →
Click here to visit Ovation Guitars Ovation Patent Drawing 1975 Click here to read a copy of the 1975 Patent for the Ovation Guitar Home Top of Pg. Read more →
J.P. Morgan Patent #8,452,703 Method and system for processing internet payments using the electronic funds transfer network. Abstract Embodiments of the invention include a method and system for conducting financial transactions over a payment network. The method may include associating a payment address of an account [...] Read more →
Modern slow cookers come in all sizes and colors with various bells and whistles, including timers and shut off mechanisms. They also come with a serious design flaw, that being the lack of a proper domed lid. The first photo below depict a popular model Crock-Pot® sold far and wide [...] Read more →
King Leopold Butcher of the Congo For the somewhat startling suggestion in the heading of this interview, the missionary interviewed is in no way responsible. The credit of it, or, if you like, the discredit, belongs entirely to the editor of the Review, who, without dogmatism, wishes to pose the question as [...] Read more →
CLAIRVOYANCE by C. W. Leadbeater Adyar, Madras, India: Theosophical Pub. House [1899] CHAPTER IX – METHODS OF DEVELOPMENT When a men becomes convinced of the reality of the valuable power of clairvoyance, his first question usually is, “How can [...] Read more →
Home Top of Pg. Read more →
Sennen Cove at Dusk – photo by Jim Champion – 2005 Home Top of Pg. Read more →
The following recipes form the most popular items in a nine-course dinner program: BIRD’S NEST SOUP Soak one pound bird’s nest in cold water overnight. Drain the cold water and cook in boiling water. Drain again. Do this twice. Clean the bird’s nest. Be sure [...] Read more →
Eadweard Muybridge was a fascinating character. Click here to learn how Eadweard committed “Justifiable Homicide” after shooting his wife’s lover in 1874. Home Top of Pg. Read more →
DECORATED or “sumptuous” furniture is not merely furniture that is expensive to buy, but that which has been elaborated with much thought, knowledge, and skill. Such furniture cannot be cheap, certainly, but the real cost of it is sometimes borne by the artist who produces rather than by the man who may [...] Read more →
“The Leda, in the Colonna palace, by Correggio, is dead-coloured white and black, with ultramarine in the shadow ; and over that is scumbled, thinly and smooth, a warmer tint,—I believe caput mortuum. The lights are mellow ; the shadows blueish, but mellow. The picture is painted on panel, in [...] Read more →
Vishnu as the Cosmic Man (Vishvarupa) Opaque watercolour on paper – Jaipur, Rajasthan c. 1800-50 CLAIRVOYANCE AND OCCULT POWERS By Swami Panchadasi Copyright, 1916 By Advanced Thought Pub. Co. Chicago, Il INTRODUCTION. In preparing this series of lessons for students of [...] Read more →
Home Top of Pg. Read more →
|